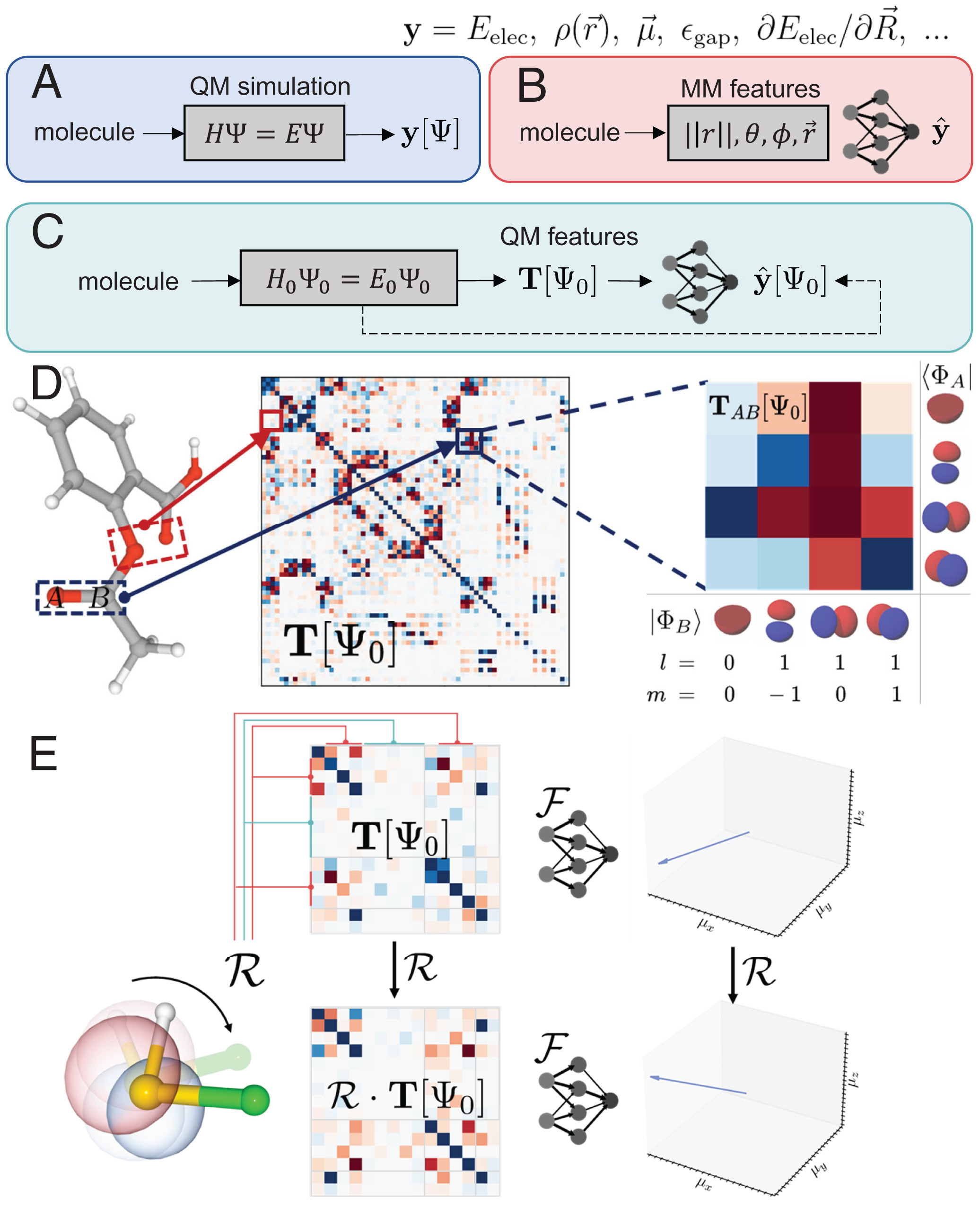
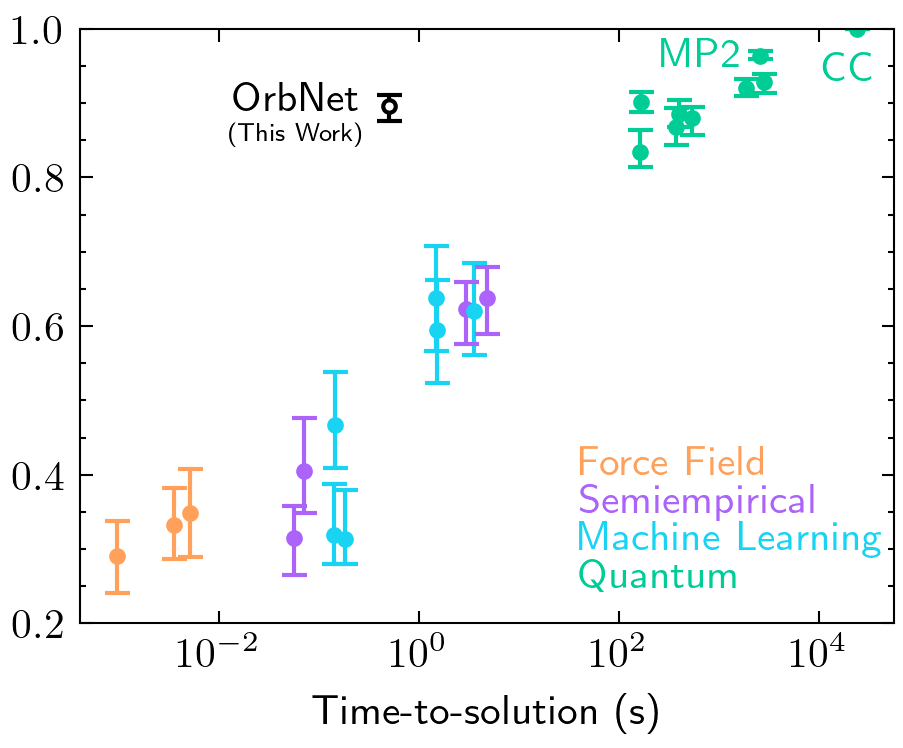
Zhuoran Qiao
Physics-Informed AI for Biochemical Discovery.

I am a Senior Machine Learning Scientist at Iambic Therapeutics, currently leading research on biomolecular structure prediction and generative AI for drug discovery. I earned my Ph.D. degree in Chemistry from Caltech CCE with a minor in Quantum Science and Engineering (Thesis), where I was fortunately advised by Prof. Anima Anandkumar and Prof. Thomas Miller. My past research centers around developing physics-driven geometric learning approaches to tackle complex problems in chemistry and structural biology.
I earned my BSc from Peking University in 2019. As an undergraduate student I worked in the Gao Group at PKU CCME, where I studied the statistical mechanics of confined soft matters.
Selected publications
 State-specific protein–ligand complex structure prediction with a multiscale deep generative modelNature Machine Intelligence 2024
State-specific protein–ligand complex structure prediction with a multiscale deep generative modelNature Machine Intelligence 2024